Prostate cancer is one of the most common cancers worldwide and also the second leading cause of cancer-related death in males in Western countries. Although the majority of human primary prostate cancers have a luminal phenotype, both basal cells and luminal cells can serve as cellular origins of prostate cancer in model systems. However, the stem cell-like plasticity of defined prostate epithelial cells and the cellular origin of prostate cancer under physiological conditions have not been identified. Recently, prostate basal and luminal cell populations were both shown to be self-sustaining, and both cell types could initiate prostate cancer. In addition, the oncogenic transformation of basal cells requires basal to luminal cell transition. These studies highlight the need to characterize the prostate cell lineage hierarchy and their behaviors in various contexts.
We are studying the prostate cell lineage hierarchy using single-cell RNA sequencing and cell lineage tracing technologies. We have identified a novel prostate luminal progenitor cell population (termed as Luminal-C) which located at the prostate the distal prostate invagination tips (termed as Dist-Luminal-C) and proximal prostate region (termed as Prox-Luminal-C). We are interested in understanding how Luminal-C cells orchestrates its niche signals in normal tissue homeostasis and diseases, and how to control the players in these machineries in vivo, as well as to provide strategies for the purpose of prostate cancer therapies.
 Luminal-C cells and their role as
prostate
luminal progenitors and cancer-initiating cells.
Luminal-C cells and their role as
prostate
luminal progenitors and cancer-initiating cells.
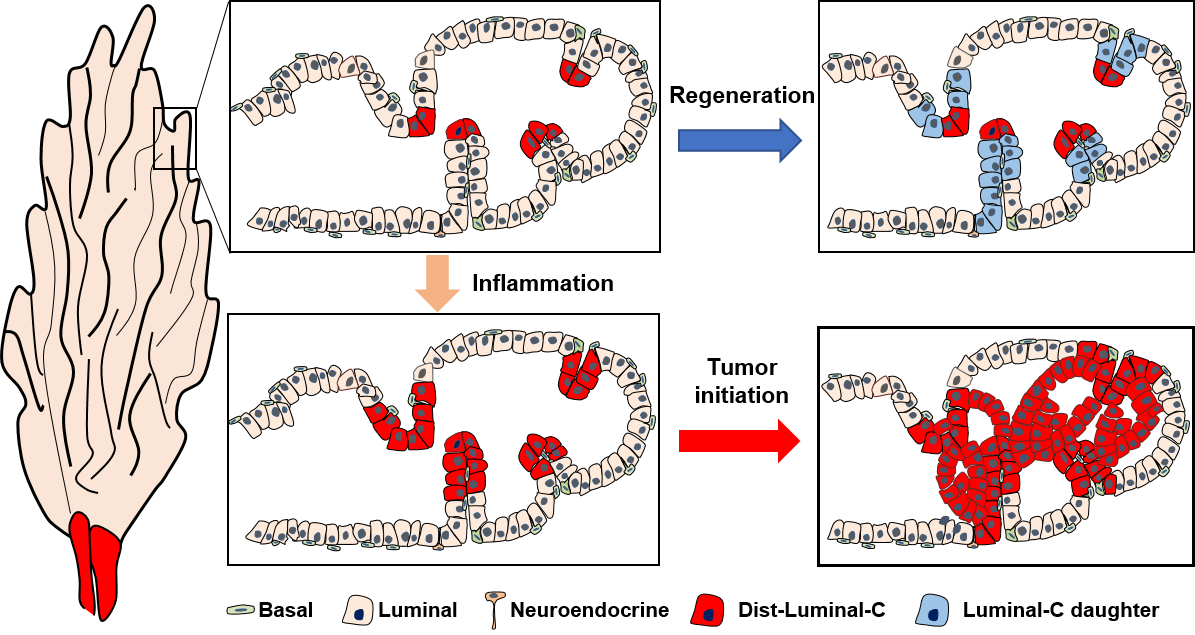
Many glandular epithelia, such as the prostate gland, consist of basal and luminal cells maintained by unipotent stem cells that can revert to multipotency during inflammation or cancer context. However, the defined basal stem cell populations responsible for prostate regeneration and their cell fates in prostate homeostasis, inflammation and carcinogenesis remain unclear. Leveraging advanced techniques including a genetic proliferation cell tracing system, enhanced lineage tracing strategies, and highly efficient basal cell labeling tools, we have demonstrated that Nkx3.1-expressing intermediate Basal-B cells have a greater tendency to generate luminal cells in vitro and under bacteria-induced prostate inflammation in vivo. Bioinformatics analysis has uncovered increased activity of the JAK/STAT signaling pathway within the Basal-B cell population. The inhibition of JAK/STAT signaling significantly diminishes the Basal-B cell fraction in normal mouse prostates and markedly hinders the basal-to-luminal differentiation capacity of Basal-B cells amidst prostate inflammation. These findings provide the JAK/STAT signaling as a potential target for patients affected by prostate inflammation associated with increased Basal-B like signature.
 Basal cell fates in prostate
inflammation.
Basal cell fates in prostate
inflammation.
Tumor initiation, progression, and therapy resistance involve genetic/epigenetic reprogramming that leads to aberrant cell lineage specification and transition. It is critical to understand the underlying mechanisms of cancer cell lineage differentiation and transition, which will provide novel insights into anticancer research. Master transcription factors have been widely recognized with the function in cell lineage transdifferentiation and cell fate reprogramming. The identification of master transcription factors in regulation cancer cell lineage specification and transition would provide tremendous insights into the mechanism of lineage plasticity in cancer progression and therapy resistance.
We are studying prostate cell lineage reprogramming at the different stages of prostate tumor initiation, progression, and therapy resistance. We have identified defined ERG as a master transcription factor to regulate prostate luminal lineage through orchestrating chromatin interactions. We are interested in understanding both plasticity of prostate cancer lineages and mechanism of therapy resistances through multi-omics analysis (RNA-seq, ATAC-seq, ChIP-seq and 3D genome analysis) and prostate cancer model analysis (Patients-derived prostate cancer organoids and genetically engineered mouse models).
 ERG drives
prostate cell fate
reprogramming through orchestrating chromatin interactions.
ERG drives
prostate cell fate
reprogramming through orchestrating chromatin interactions.
Prostate cancer adeno-to-neuroendocrine lineage transition has emerged as a mechanism of targeted therapeutic resistance. Identifying the direct molecular drivers and developing pharmacological strategies using clinical-grade inhibitors to overcome current lineage transition-induced therapeutic resistance are imperative. Using single-cell multiomics analyses, we have investigated the dynamics of cellular heterogeneity, transcriptomics regulation and microenvironmental factors in mouse prostate cancer samples with complete time series of tumor evolution seen in patients. We identified FOXA2 orchestrated prostate cancer adeno-to-neuroendocrine lineage transition, and FOXA2 expression was significantly induced by androgen deprivation. KIT pathway was directly regulated by FOXA2 and specifically activated in neuroendocrine prostate cancer (NEPC). Pharmacologic inhibition of Kit signaling significantly suppressed mouse and human NEPC tumor and organoids growth. These findings reveal that Foxa2 drives adeno-to-neuroendocrine lineage plasticity in prostate cancer, and provide a potential pharmacological strategy for castration-resistant NEPC. This study provides a high-resolution transcriptional and chromatin accessibility landscape of prostate cancer luminal-to-neuroendocrine lineage transition, reveals critical roles of Foxa2 and Foxa1 in mediating prostate luminal-to-neuroendocrine lineage transition and the activation of Kit signaling in NEPC, and highlight KIT signaling as a potential therapeutic target for NEPC.
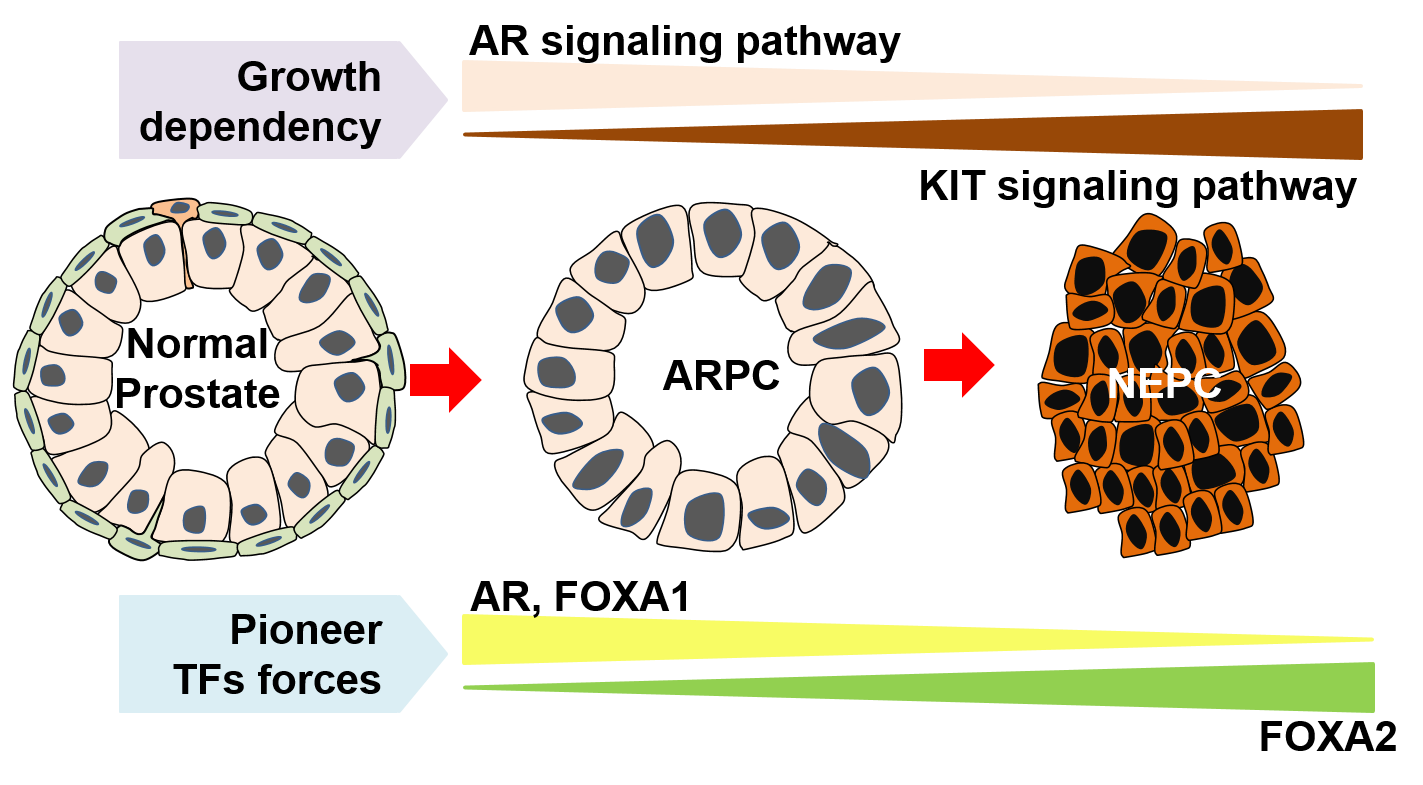 FOXA2 drives lineage plasticity
and KIT pathway
activation in neuroendocrine prostate cancer.
FOXA2 drives lineage plasticity
and KIT pathway
activation in neuroendocrine prostate cancer.
Epigenetic regulation profoundly influences the fate of cancer cells and their capacity to switch between lineages by modulating essential gene expression, thereby shaping tumor heterogeneity and therapy response. Utlizing single-cell RNA-sequencing (scRNA-seq) on both human and mouse prostate cancer samples, combined with whole-genome bisulfite sequencing (WGBS) and multiple genetically engineered mouse models (GEMMs), we investigated the molecular mechanism of AR independent lineage plasticity and uncovered a potential therapeutic strategy. Single cell transcriptomic profiling of human prostate cancers, both pre- and post-androgen deprivation therapy (ADT), revealed an association between Liver kinase B1 (LKB1) pathway inactivation and AR-independence. LKB1 inactivation led to AR-independent lineage plasticity and global DNA hypomethylation during prostate cancer progression. Importantly, the pharmacological inhibition of DNA hypomethylation and supplementation with metabolite were found to effectively suppress AR-independent prostate cancer growth. These insights shed light on the mechanism driving AR independent lineage plasticity and propose a potential therapeutic strategy by targeting DNA hypomethylation in AR-independent CRPC.
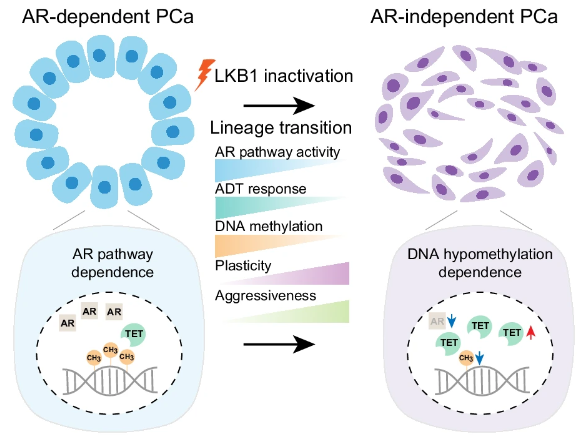 LKB1 inactivation promotes
epigenetic
remodeling-induced lineage plasticity and antiandrogen resistance in prostate
cancer.
LKB1 inactivation promotes
epigenetic
remodeling-induced lineage plasticity and antiandrogen resistance in prostate
cancer.
Sex differences are prevalent in human development, physiology, and disease. Phenotypic sex differences are shaped by the combined effects of endogenous factors and exogenous environmental exposures. Hormones, especially androgens and estrogens, are thought to contribute substantially to sex-biased phenotypes. The actions of androgens are mediated by binding to androgen receptor (AR), a ligand-dependent nuclear transcription factor. Drugs targeting AR as well as androgen synthesis to inhibit the androgen pathway, including enzalutamide and abiraterone, have brought great benefits to prostate cancer patients. These studies highlight the need to systematically explore how androgens modulate sex differences and to investigate the application potential of targeting the androgen pathway for sex-biased diseases.
We have constructed a high-dimensional single-cell transcriptomic atlas comprising over 2.3 million cells from 17 tissues in Mus musculus and explored the impacts of sex and androgens on the molecular programs and cellular populations. In particular, we found that sex-biased immune gene expression and immune cell populations, such as group 2 innate lymphoid cells (ILC2s), were modulated by androgens. Integration with the UK Biobank dataset revealed potential cellular targets and risk gene enrichment in antigen presentation for sex-biased diseases. This study lays the groundwork for understanding the sex differences orchestrated by androgens and provides important evidence for targeting the androgen pathway as a broad therapeutic strategy for sex-biased diseases.
We developed a web tool (https://casadbtools.com/) to provide customized visualization and analyses of our scRNA-seq dataset.
 A web tool to
provide customized
visualization and analyses of sex differences at single-cell resolution.
A web tool to
provide customized
visualization and analyses of sex differences at single-cell resolution.
The lack of in vitro cancer models that recapitulate the diversity of human cancers has hampered progress in understanding cancer cell lineage transition and therapy response. Organoid technology has been remarkably improved in recent years to allow the rapid generation of large repertoire of patient-derived cancer organoids (PDOs) amenable to genetic and pharmacologic studies. PDOs have been used to elucidate crucial scientific questions, including the relationships between genetic/epigenetic alterations and drug responses, cell plasticity during disease progressions, and mechanisms of drug resistances. We have generated patient-derived organoid biobanks of prostate, pancreatic, lung and liver cancers. These tools will help us address the cell origin of cancers, mechanistically study cancer cell lineage transition and therapy response.
For centuries, the attempts have been continuously made to artificially reconstitute counterparts of the in vivo organs from their tissues or cells. Only in the recent decade, organoid technology as a whole technological field systematically has emerged and been shown to play important roles in tissue engineering.
The liver hepatobiliary architecture is critical and essential for proper hepatic function, highlighting the need for an in vitro liver model with the real histological characteristics to study liver diseases and regeneration. We are developing a novel culture system of liver organoids to generate hepatobiliary architecture with hepatic function in vitro and upon transplantation in vivo.
Chinese Academy of Sciences Building C, Room 307
320 Yue Yang Road, Xu Hui District, Shanghai, China 200031
E-mail:Dong.gao[at]sibcb.ac.cn;
Office Tel: (+86) 21-54921117;
Lab Manager Tel: (+86) 21-54921103